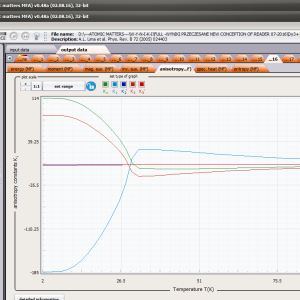
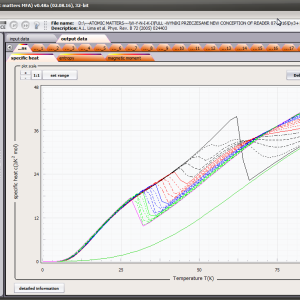
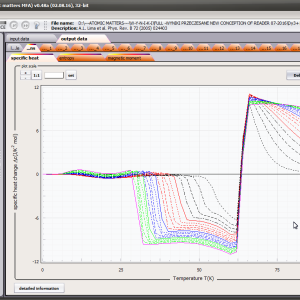
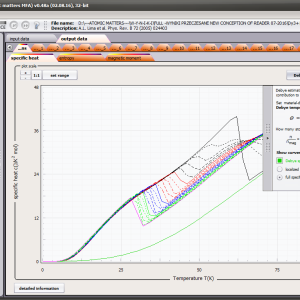
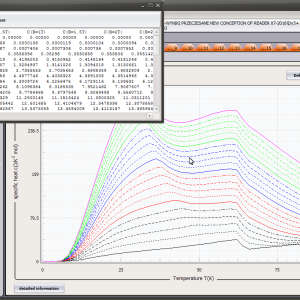
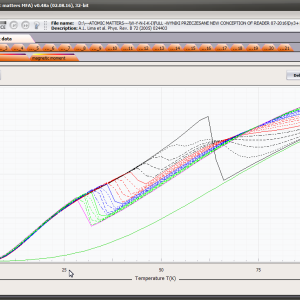
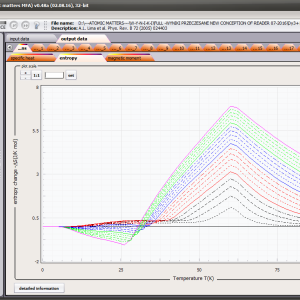
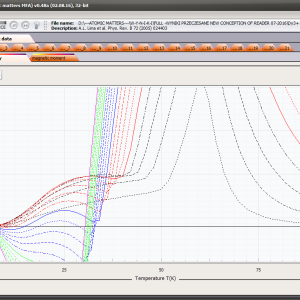


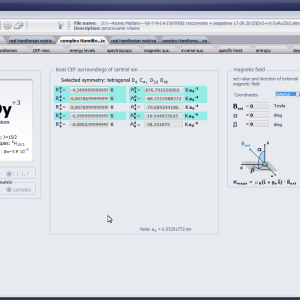
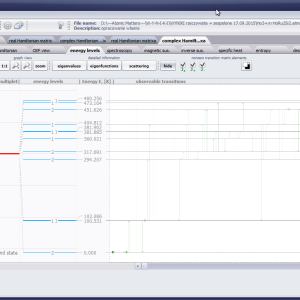
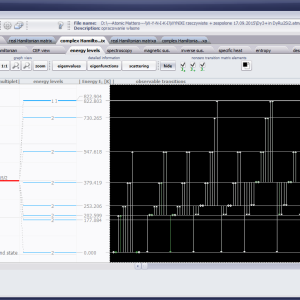
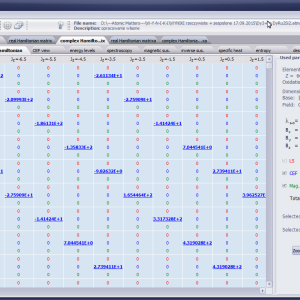
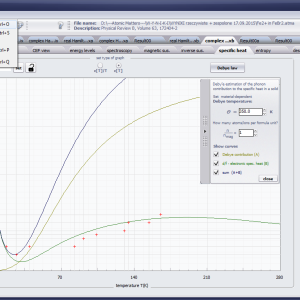
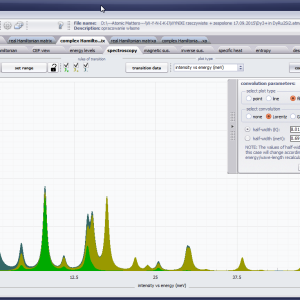
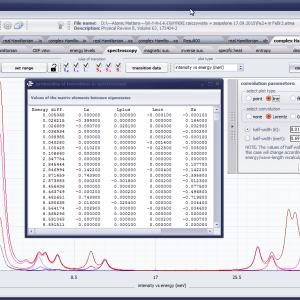
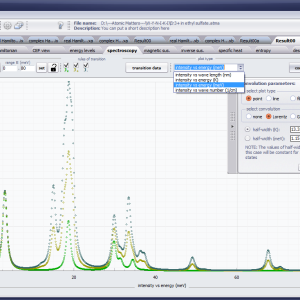
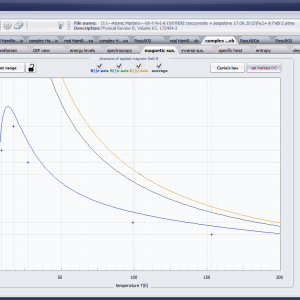
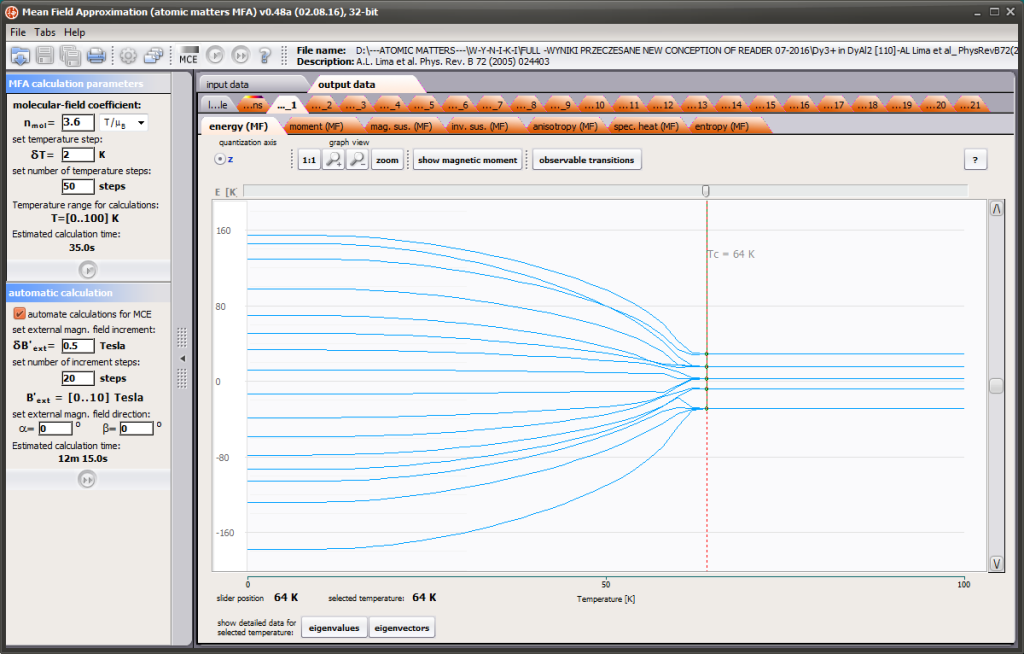
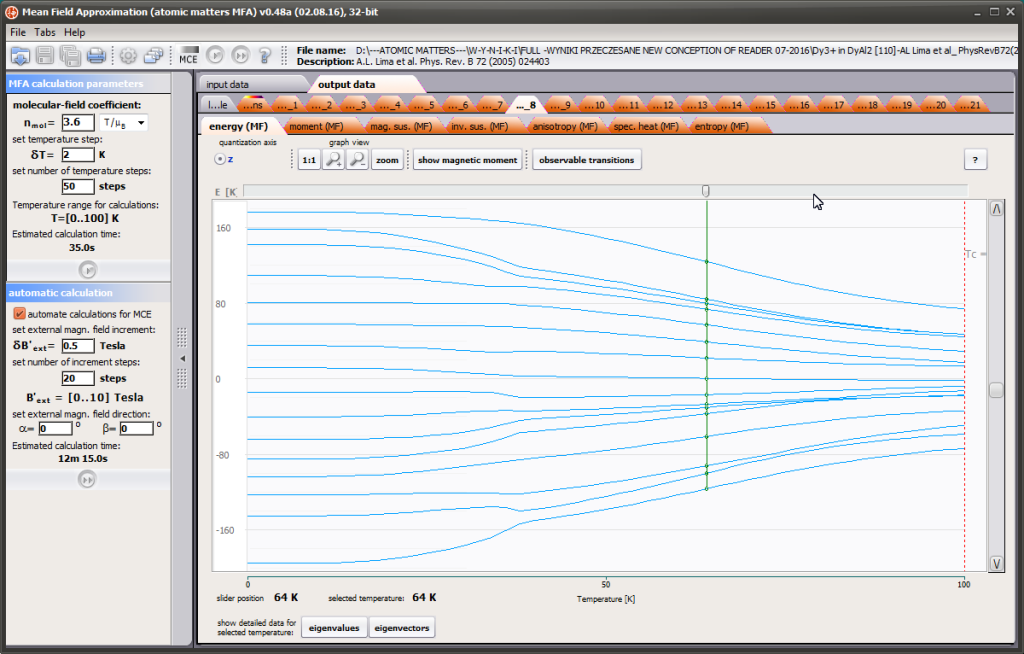
LIMITATIONS OF APPLICATION
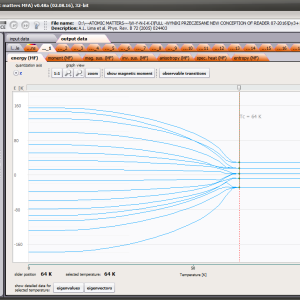
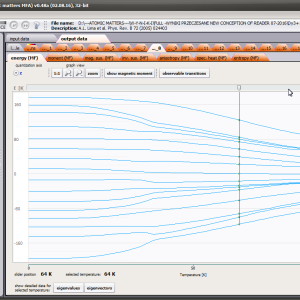
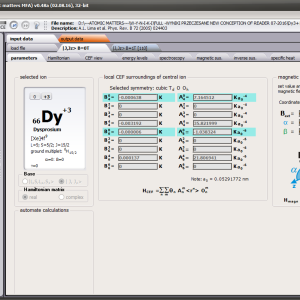
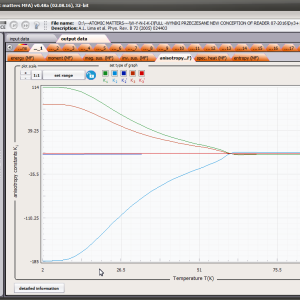
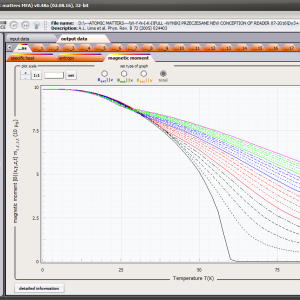
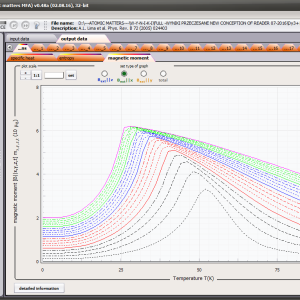
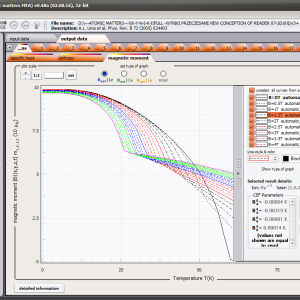
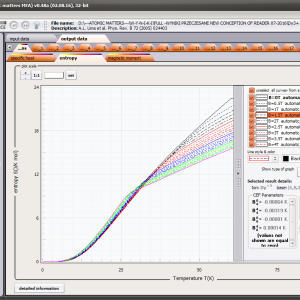
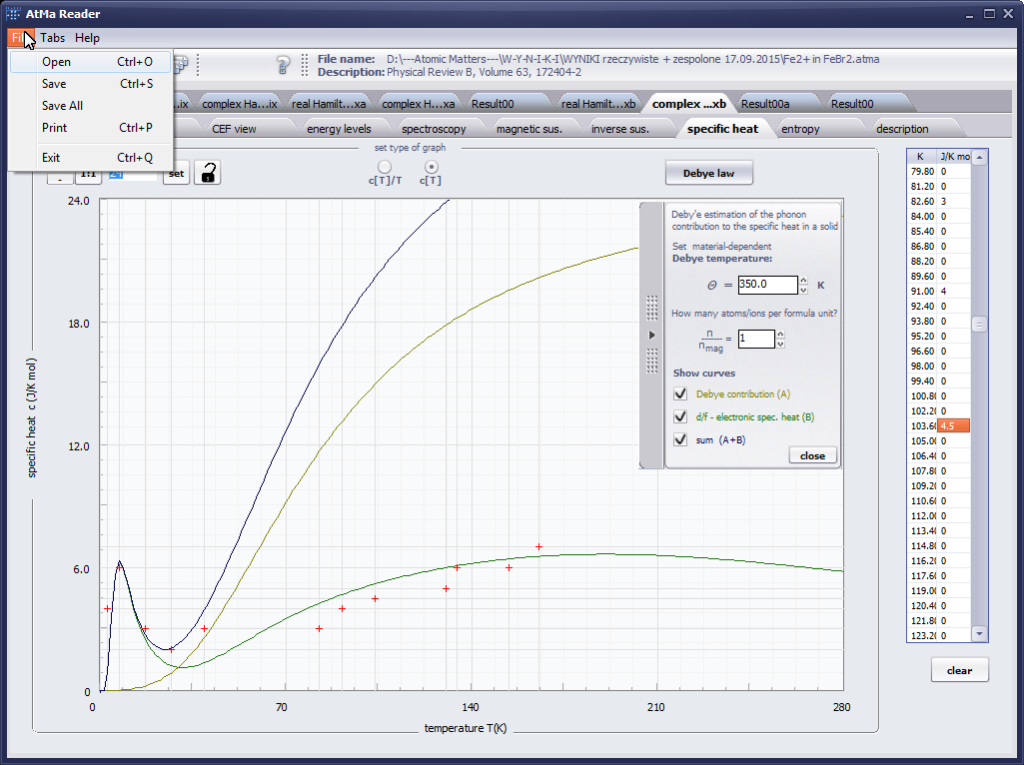
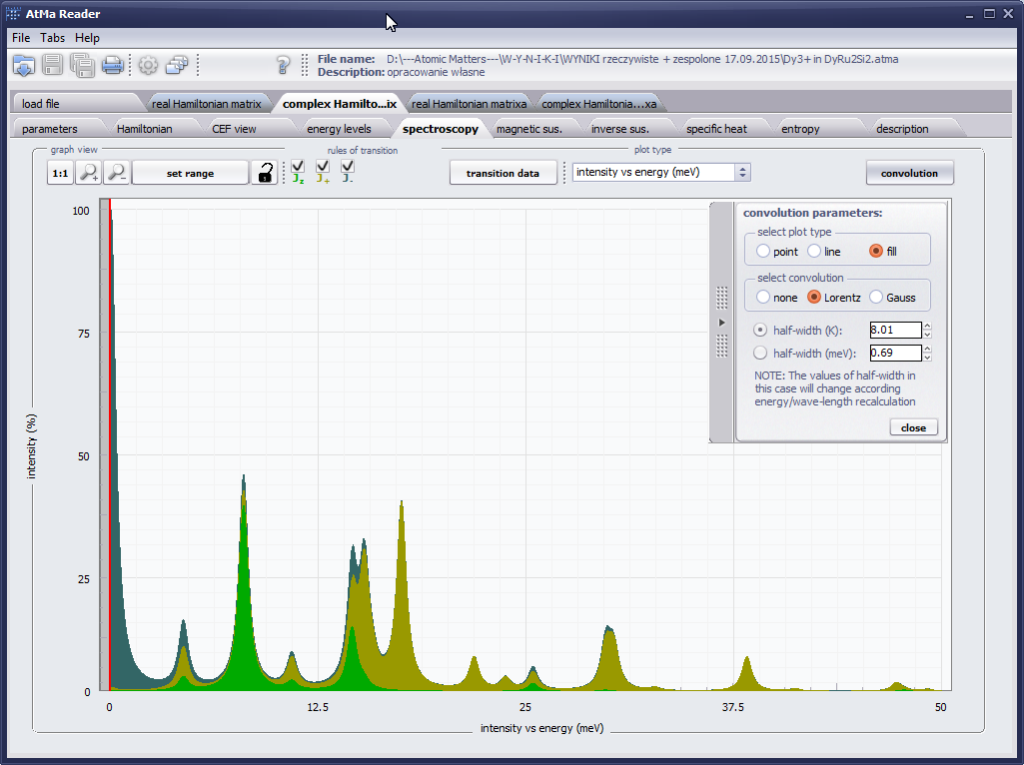
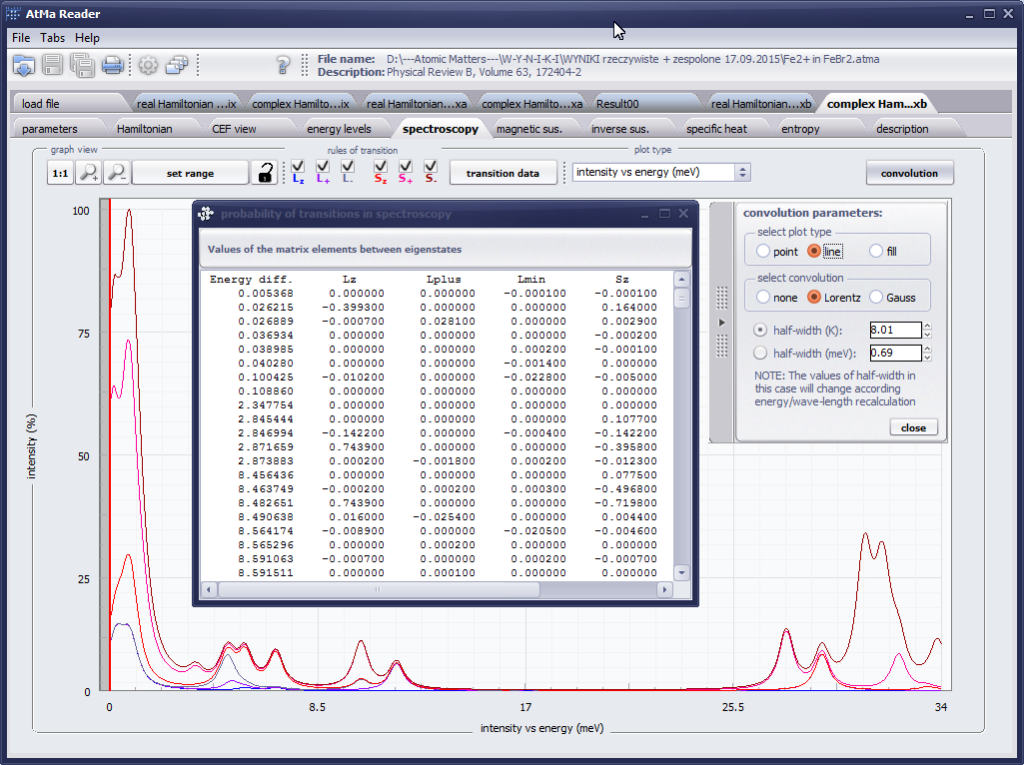
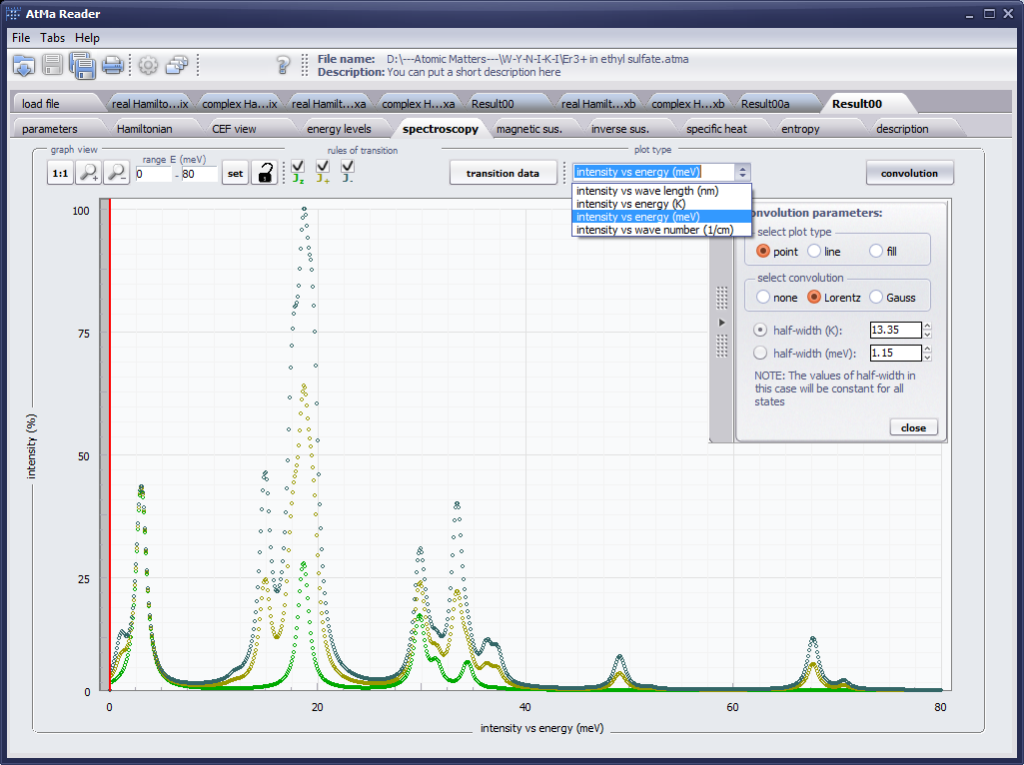
The only methodological limitation is the condition of locating multi-electron states on an atom or ion . This does not mean the need for modeling systems with ionic bonds or, as is often wrongly assumed, insulators. Computational practice has shown a very high ability to predict the properties in the case of intermetallic compounds. The above restriction should therefore be understood as the need to preserve the integrity of shell p, d or f - that is, the assumption of said electrons not participating in transport phenomena (conductivity). Of course, the software has a number of IT-related limitations, the most important which is the modeling of systems containing only one component with an unclosed shell p, d or f. Of course, in the case of modeling crystals with a larger number of various paramagnetic ions, you can separately simulate various network positions for different ions/atoms but their effect on each other in this version of the software can only be simulated by setting permanent external fields with defined symmetry (without self-alignment). The conference discussions on theoretical solid state physics, disputes often arise regarding the appropriateness of using a particular theoretical approach to the issues of describing the properties of a particular group of compounds. By providing this application we want to give a chance for a strict, non-doctrinal definition of the applicability of computational methods referred to above. The authors are aware, however, that single-ion crystal characteristics play an important, although not the only role in the properties of the crystal. The effects associated with the domain structure and long-range interactions, dimerization or possible delocation are not taken into account in this approach. In particular, it is worth remembering that the issues of preparation of actual materials (such as permanent magnets) require creating a higher scale material structures and their theoretical description requires special methodology taking into account these structures. However, magnetocrystalline single-ion anisotropy is the basic contribution that allows building models of domains, crystallites, sinters etc. The results of single-ion calculations allow determining the extreme values of magnetocrystalline parameters, providing grounds for e.g. the search for the maximum value of saturation in a given direction in the crystal.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
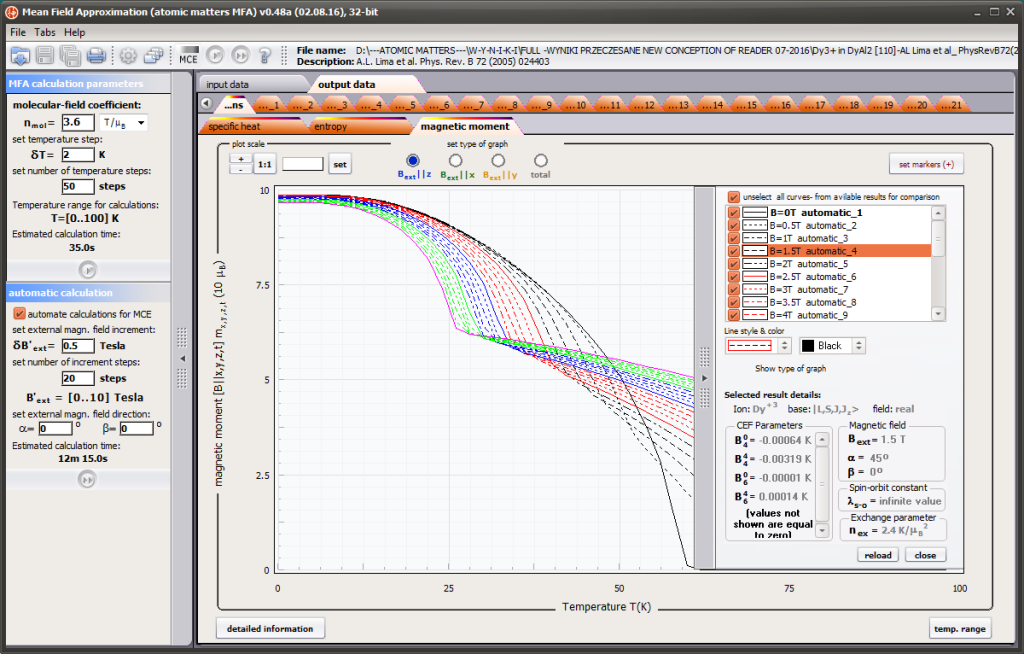{kind=link}
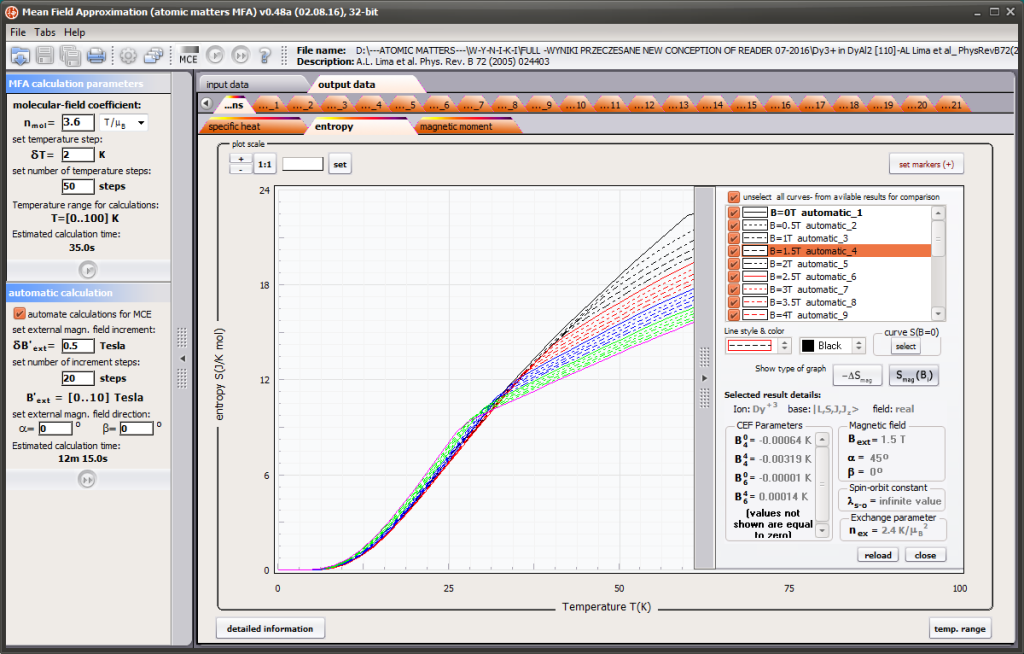{kind=link}
{kind=link}
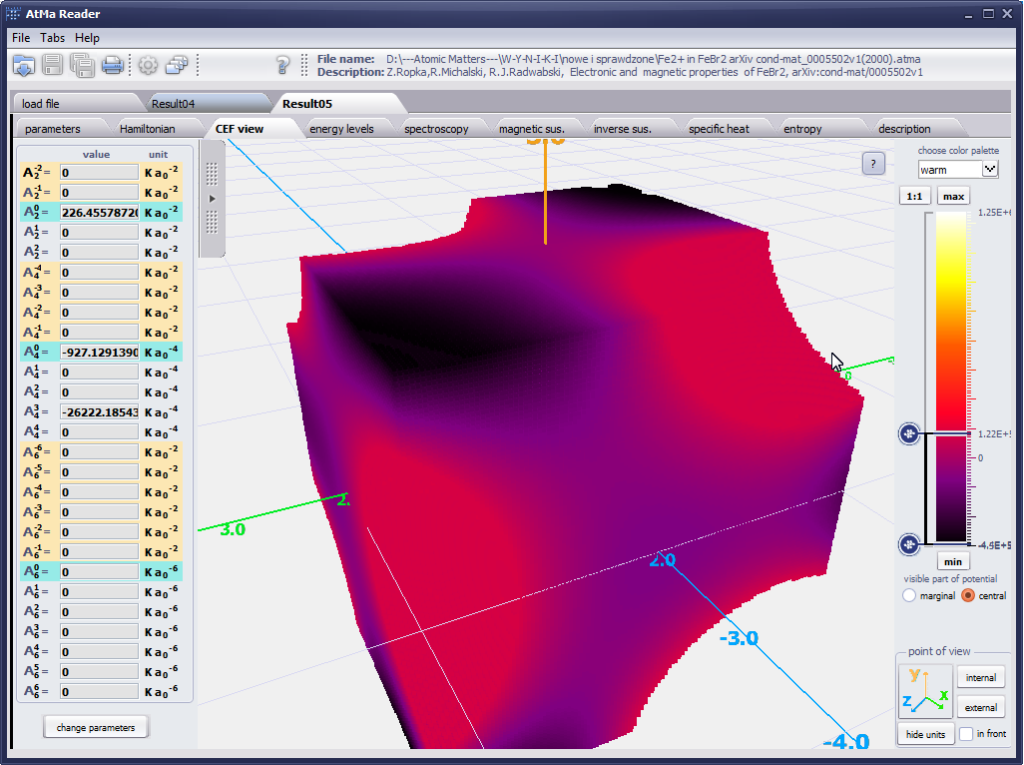{kind=link}
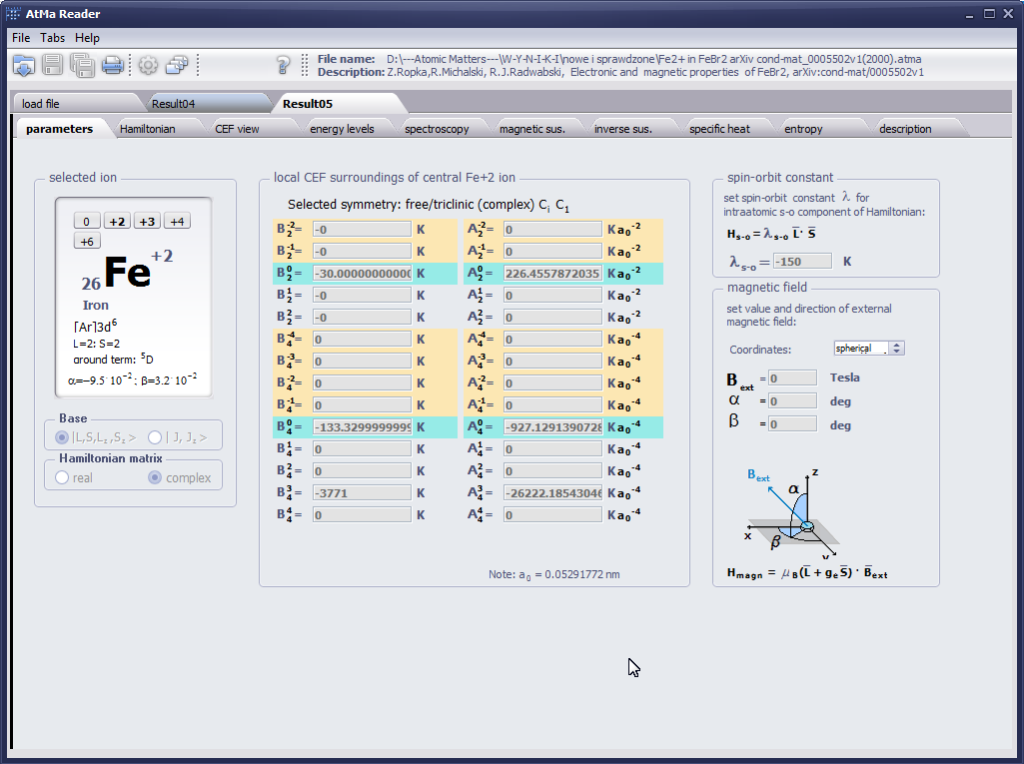{kind=link}
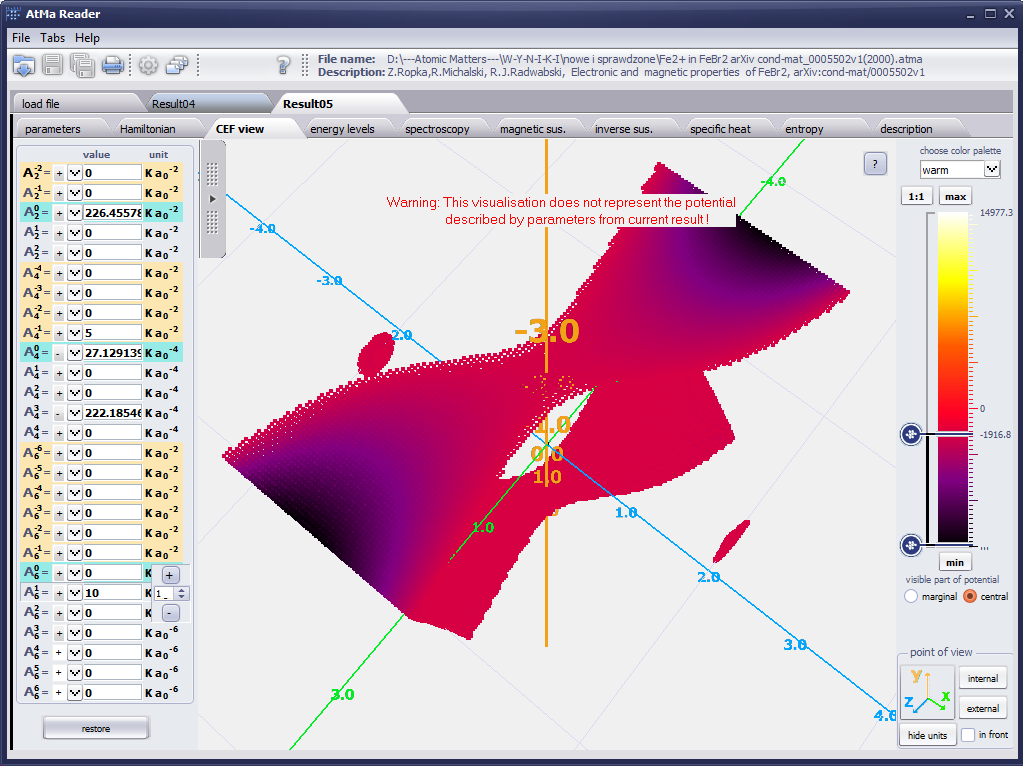{kind=link}
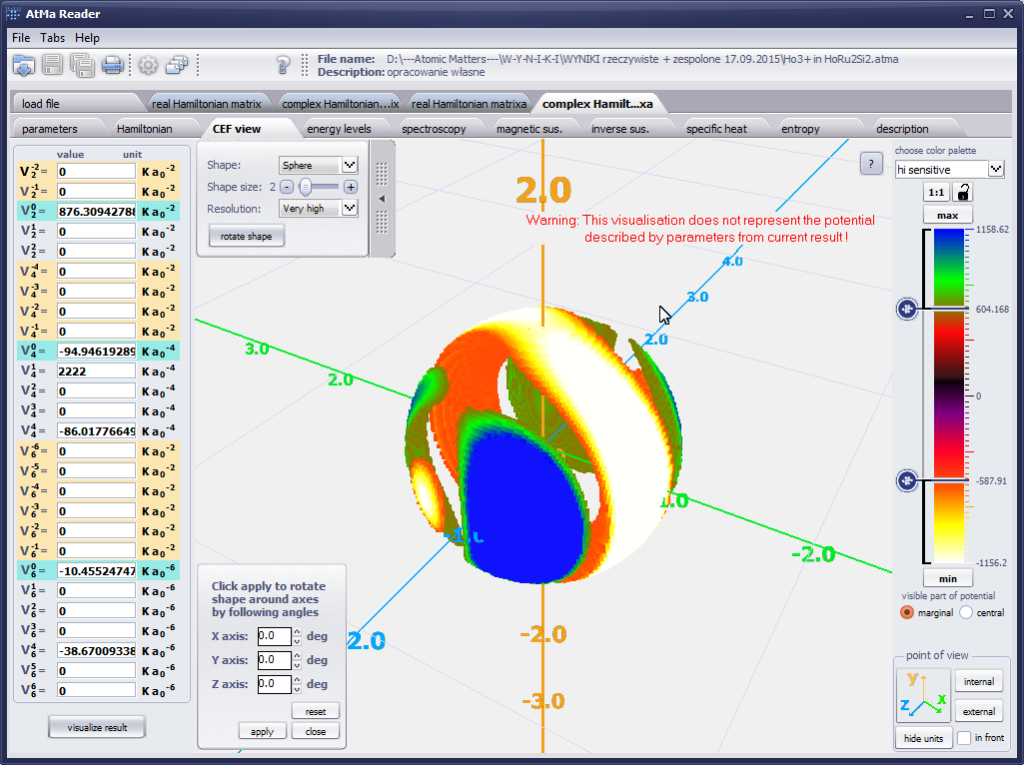{kind=link}
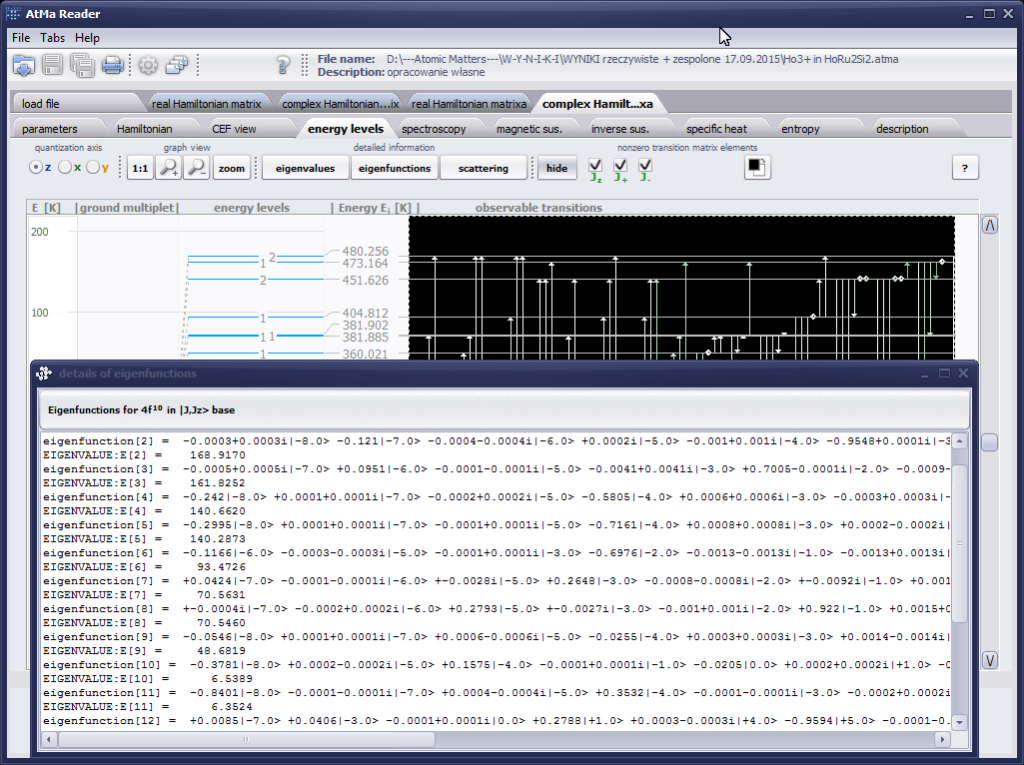{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}