
Aplikacja ATOMIC MATTERS jest unikalnym, tworzonym przez fachowców nauk ścisłych narzędziem umożliwiającym możliwie najpełniejszy i najściślejszy opis właściwości fizycznych atomowych układów elektronowych poddanych oddziaływaniom elektromagnetycznym, typowym dla sytuacji gdy na niezamkniętą powłokę elektronową oddziałuje potencjał elektrostatyczny o definiowalnej symetrii oraz pole magnetyczne. Przez przeszło dekadę tworzyliśmy przyjazne użytkownikowi oprogramowanie pozwalające w ścisłym kwantowo-mechanicznym rygorze modelować te właściwości materii, których natura ma charakter jednojonowy (jednoatomowy). Metody obliczeniowe aplikacji ATOMIC MATTERS, opracowane w oparciu o własne procedury operacji generowania i diagonalizacji macierzy (operatorów ekwiwalentnych) o elementach zespolonych są w ścisłej zgodności z konwencją i metodologią współczesnej fizyki atomowej, fizyki ciała stałego i technikami numerycznymi opisywanymi w powszechnie poważanych środowiskowo, publikacjach naukowych. Konsekwentnie dążąc do maksymalnego uproszczenia interface’u aplikacji osiągnęliśmy możliwość prowadzenia rzetelnego procesu badawczego w obszarze właściwości materiałów magnetycznych i prognozowania właściwości związków stałych bez znajomości zawiłości związanych z aksjomatyką, formalizmem i metodologią mechaniki kwantowej. Dzięki zaimplementowanej logice działania aplikacji, możliwe jest prognozowanie właściwości magnetycznych cieplnych i elektronowych w zakresie całych szeregów izostrukturalnych. Oznacza to, że ATOMIC MATTERS dostarcza możliwości przewidywania tych właściwości fizycznych materiałów, które mają naturę pochodzącą od zlokalizowanych stanów elektronowych. Z praktycznego punktu widzenia, rola tych stanów elektronowych dla właściwości magnetycznych, spektralnych i kalorymetrycznych w przypadku materiałów zawierających metale przejściowe, metale ziem rzadkich, uranowce jest absolutnie fundamentalna.
ALGORYTM DZIAŁANIA APLIKACJI ATOMIC MATTERS
Schemat blokowy interakcji z narzędziem, prowadzący od zdefiniowania przedmiotu obliczeń do uruchomienia obliczeń i uzyskania zestawu wyników (drzewo decyzyjne użytkwnika) przedstawia diagram poniżej.
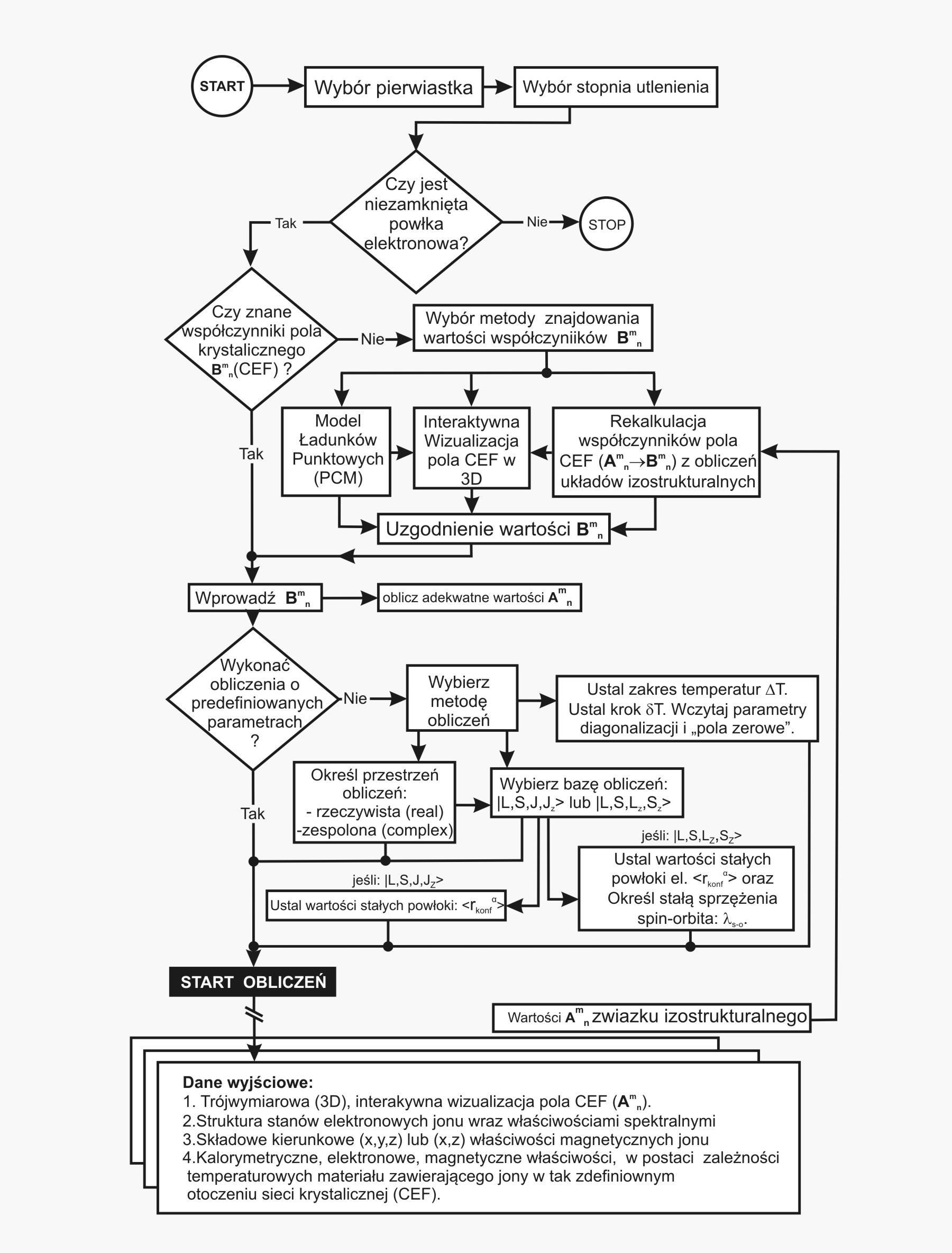
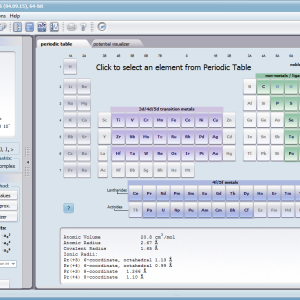
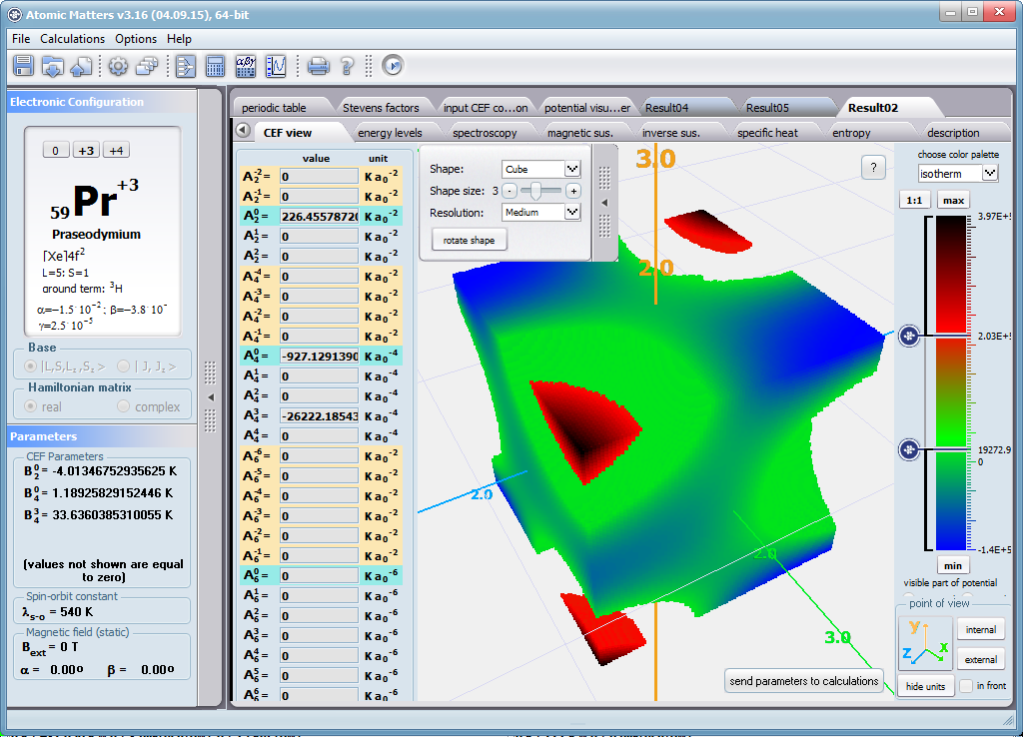
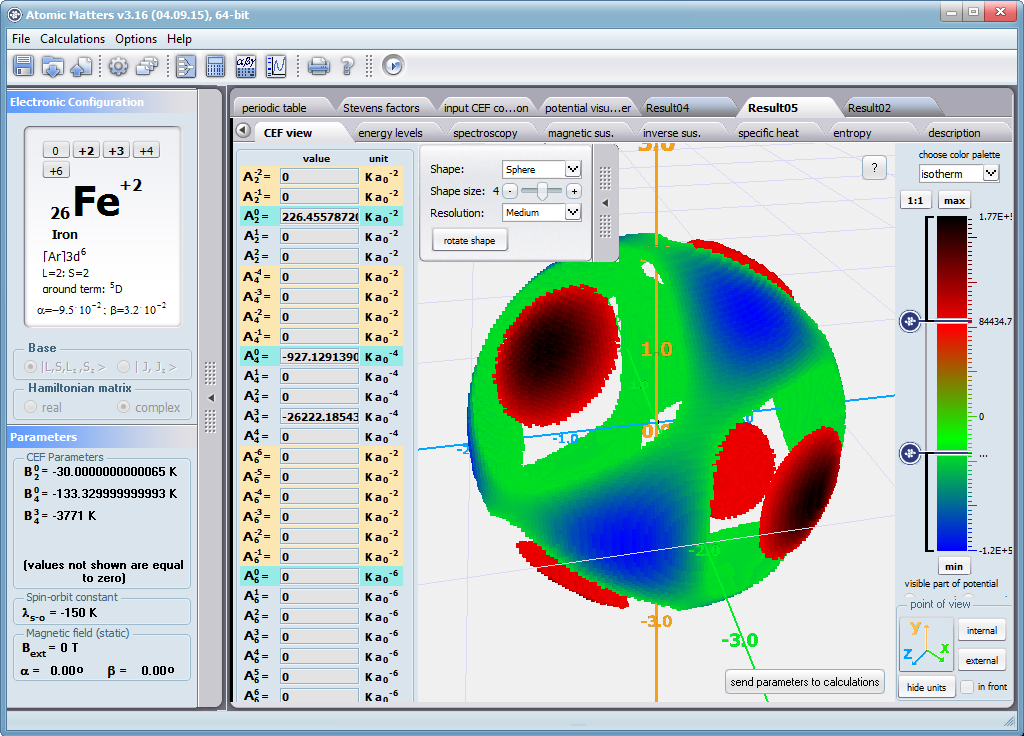
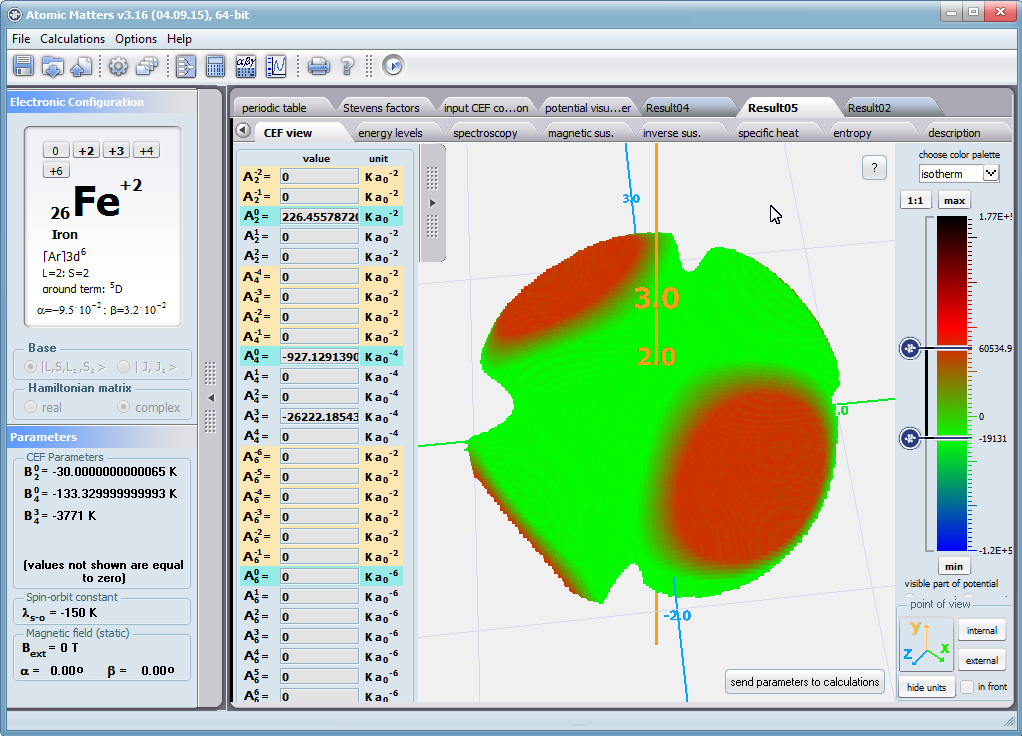
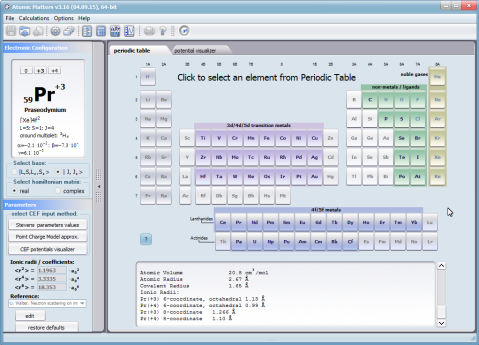
Dzięki użyciu najnowocześniejszych metod numerycznych do diagonalizacji zespolonych macierzy hamiltonianów oddziaływań elektronów symulowanego jonu/atomu osiągnęliśmy prędkość działania programu pozwalającą na uzyskanie pełnego zestawu wyników obliczeniowych (dla obliczeń typowych, bez narzucenia cykliczności pracy) w czasie rzędu sekund na sprzęcie komputerowym PC o niewygórowanych parametrach. Sposób pracy aplikacji ATOMIC MATTERS polega na generowaniu kompletnych, zwizualizowanych w postaci wykresów, diagramów i projekcji 3D kompletów danych wejściowo- wyjściowych na zestawach zakładek o intuicyjnej, hierarchicznej strukturze. Przedstawiona jest ona wizualnie w postaci warstw zakładek grupujących serie wyników. Pozwala to na szybką weryfikację wpływu określonego oddziaływania na strukturę jonu/atomu oraz wpływu tej zmiany na właściwości makroskopowej próbki materiału. Operując na całych zestawach danych wyjściowych i wejściowych łączących geometrię otoczenia jonu paramagnetycznego w sieci krystalicznej z właściwościami makroskopowego kryształu idealnego, składającego się z takich jonów/atomów oraz jonów/atomów zamkniętopowłokowych, nie wnoszących wkładu paramagnetycznego, symulujemy te właściwości materiału realnego, które mają naturę najbardziej pierwotną. ATOMIC MATTERS to narządzie, dzięki któremu można szybko i efektywnie symulować wpływ pola elektrycznego określonego przez szereg potencjałów multipolowych (kwadrupola, oktupola etc.) o zdefiniowanych symetriach lokalnych oraz stałego pola magnetycznego o zadanym kierunku w przestrzeni, na jon/atom z niezamkniętą powłoką (czyli tzw. jon/atom paramagnetyczny, jon/atom metali przejściowych), dowolnie wybrany z Układu Okresowego Pierwiastków (tablicy Mendelejewa). W aplikacji, obszary dostępnych do prowadzenia obliczeń pierwiastków w Układzie Okresowym są wyróżnione kolorystycznie na zakładce pojawiającej się po uruchomieniu aplikacji. Wybór innych pierwiastków na tej zakładce pozwala na dostęp do ważniejszych informacji zebranych pod kątem użyteczności dla prowadzenia obliczeń w obszarze ustalenia geometrii ligandów w związkach koordynacyjnych. Teoria i matematyczne metody obliczeniowe związane z rozkładem pola elektrycznego otaczającego jon paramagnetyczny wywodzące się z fizyki atomowej i teorii grup powstały i rozwijały się intensywnie w drugiej połowie XX w. Zawiłość rachunku operatorowego dedykowanego dla przestrzeni definiowanej przez klasy symetrii pola ligandów utrudniała rozwój metod obliczeniowych które z czasem wytworzyły uproszczone narzędzia kalkulacyjne (np. diagramy LLW – Lea, Leask , Wolf, lub wzory de Gennes’a). Trudności związane z definiowaniem i weryfikacją potencjału pola krystalicznego CEF jonu/atomu i jego związkiem ze strukturą krytaliczną kryształu rozwiązaliśmy przez interaktywną wizualizację potencjału koordynującego z licznymi narzędziami pozwalającymi regulować paletę barw i jej związek z wartością potencjału, widzialność (potencjały ekstremalne, przyzerowe), figurę projekcyjną(sześcian/kula) pozycję obserwatora (wewnątrz, zewnątrz, zoom) itp.Schemat blokowy obliczeń wykonywanych w podczas jednego cyklu zwykłego działania aplikacji ATOMIC MATTERS:
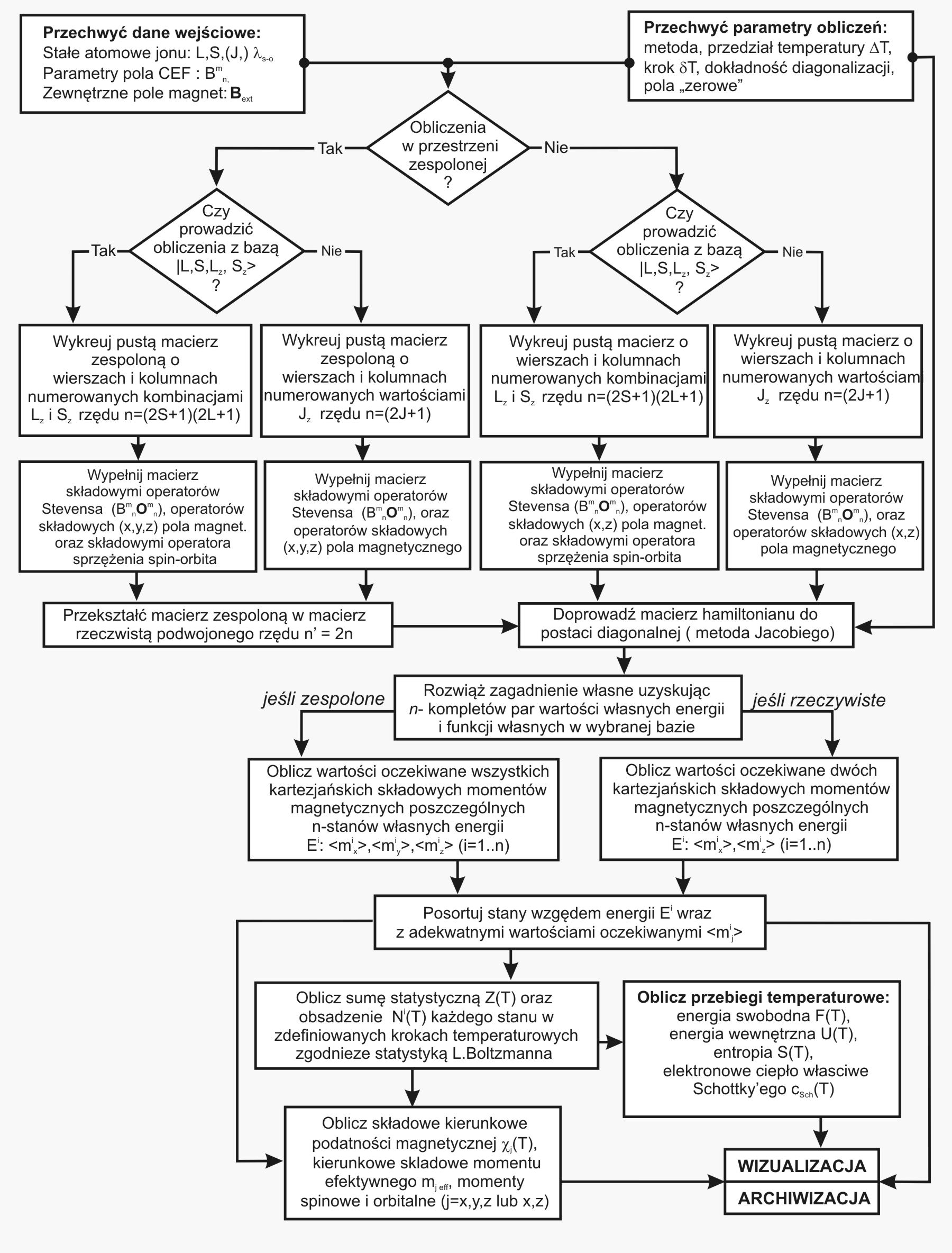
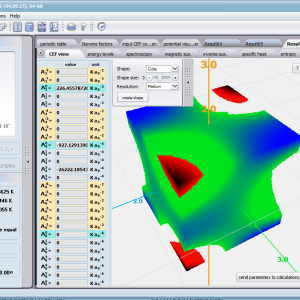
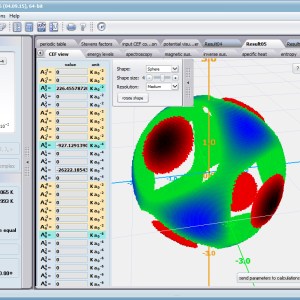
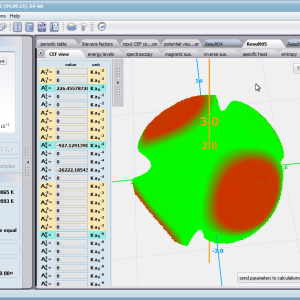
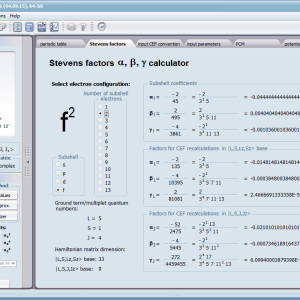
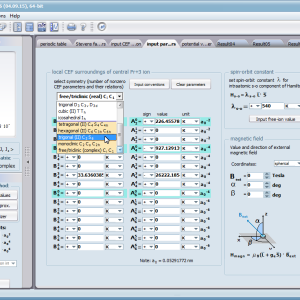
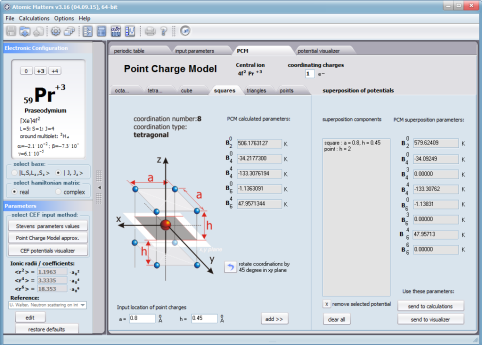
Aplikacja ATOMIC MATTERS pozwala oglądać i dowolnie składać potencjały rozwinięcia szeregu multipolowego (w postaci harmonik tesseralnych, później realizowanych obliczeniowo w wielowlowymiarowej przestrzeni krętowych liczb kwantowych za pomocą języka operatorów wprowadzonych do f.c.s przez K.W.H. Stevensa – patrz podstawy teoretyczne), w którym, zgodnie z regułami fizyki atomowej możesz umieścić wybrany jon paramagnetyczny i po uruchomieniu obliczeń (okrągły przycisk na belce programu) uzyskać zestaw wyników obliczeniowych dla tak zdefiniowanego kryształu (w funkcji temperatury na zadanym jej przedziale). Operacja umieszczania różnych jonów/atomów w tym samym potencjale pozwala względnie skutecznie symulować własności całych szeregów związków (oczywiście, przy ich względnej izostrukturalności, przynajmniej w zakresie bezpośredniego otoczenia jonu/atomu paramagnetycznego).
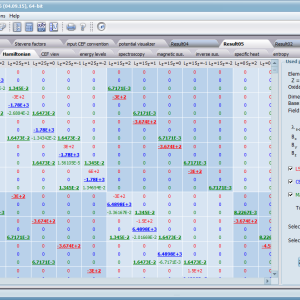
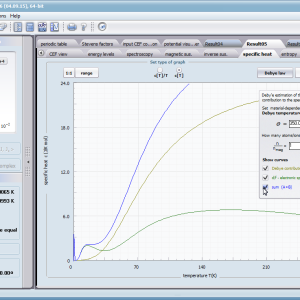
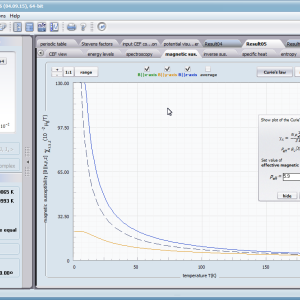
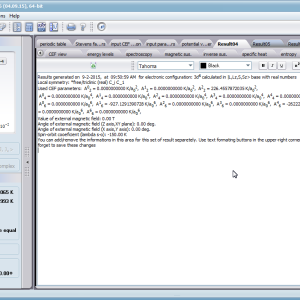
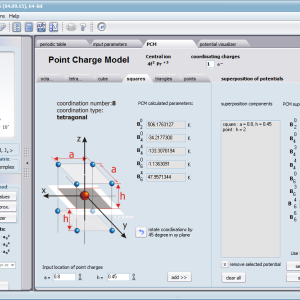
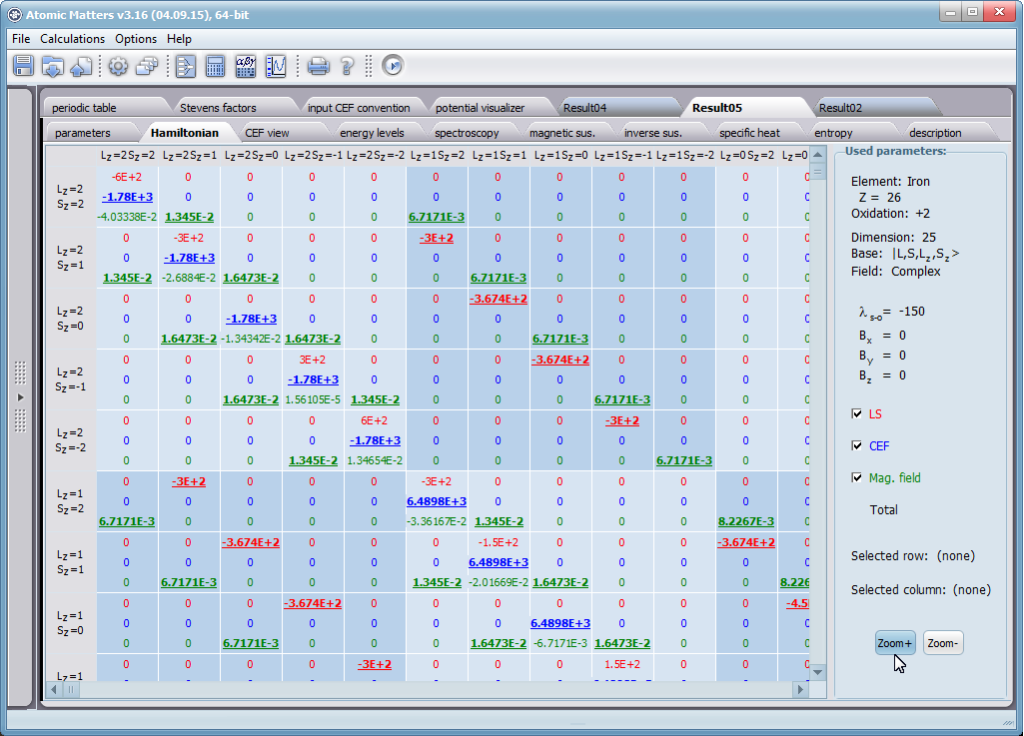
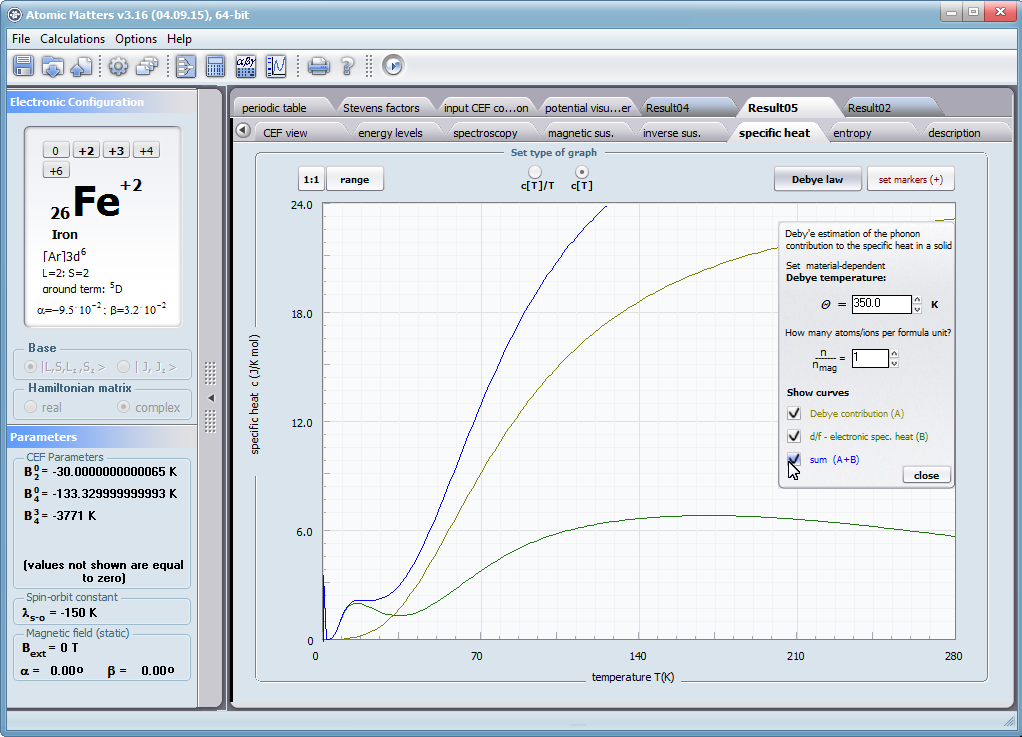
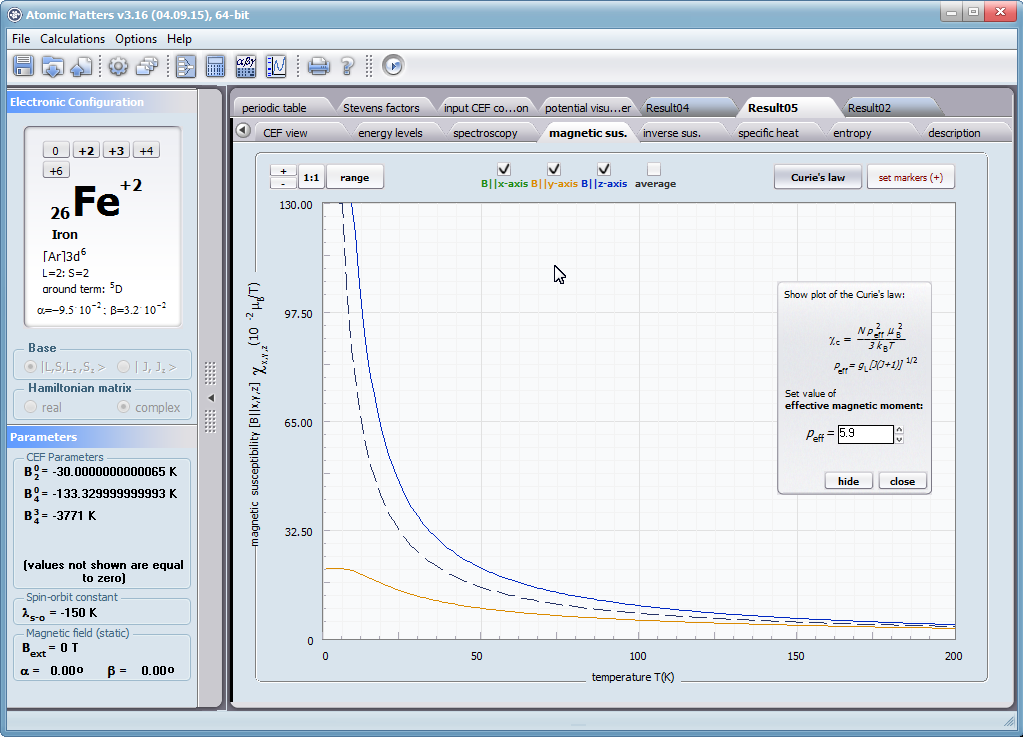
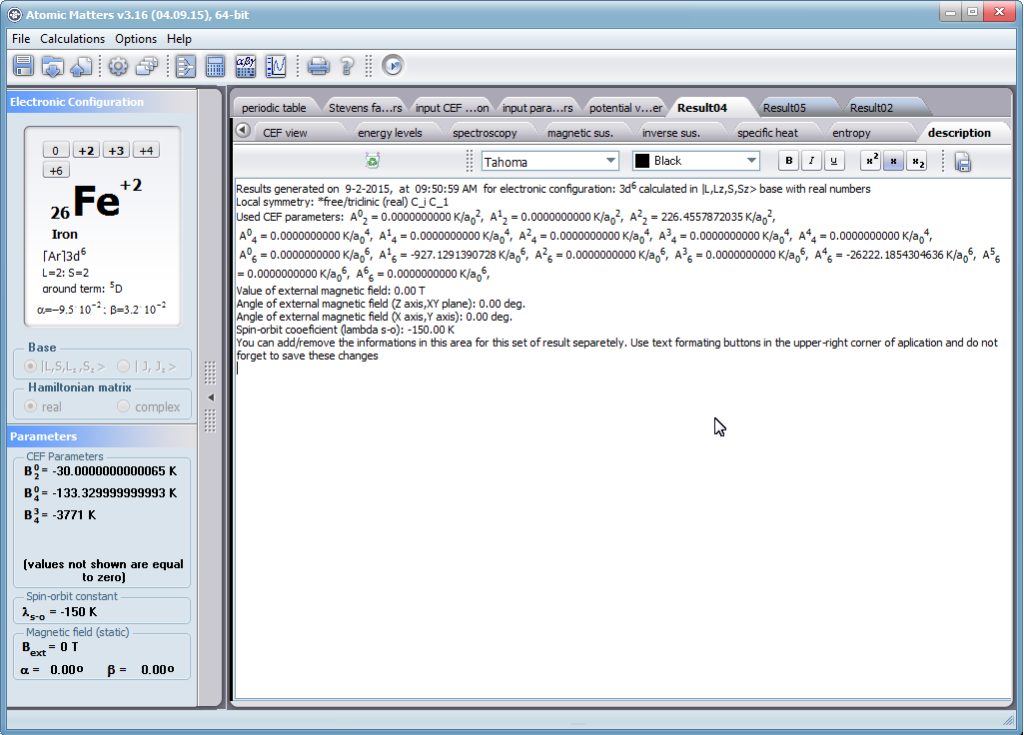
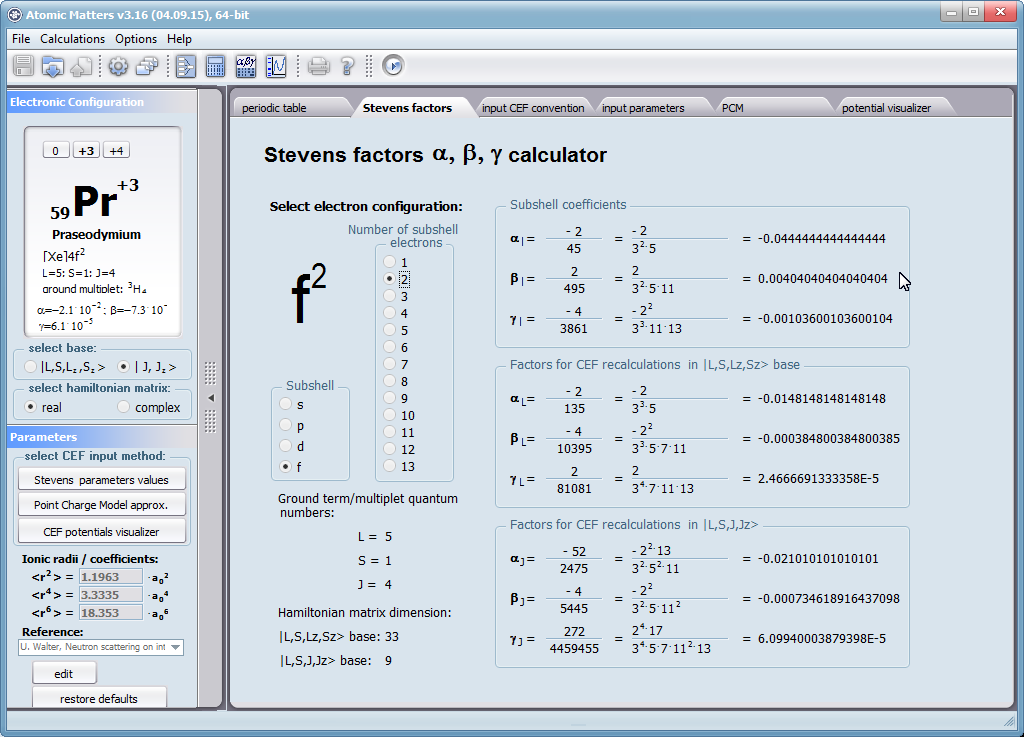
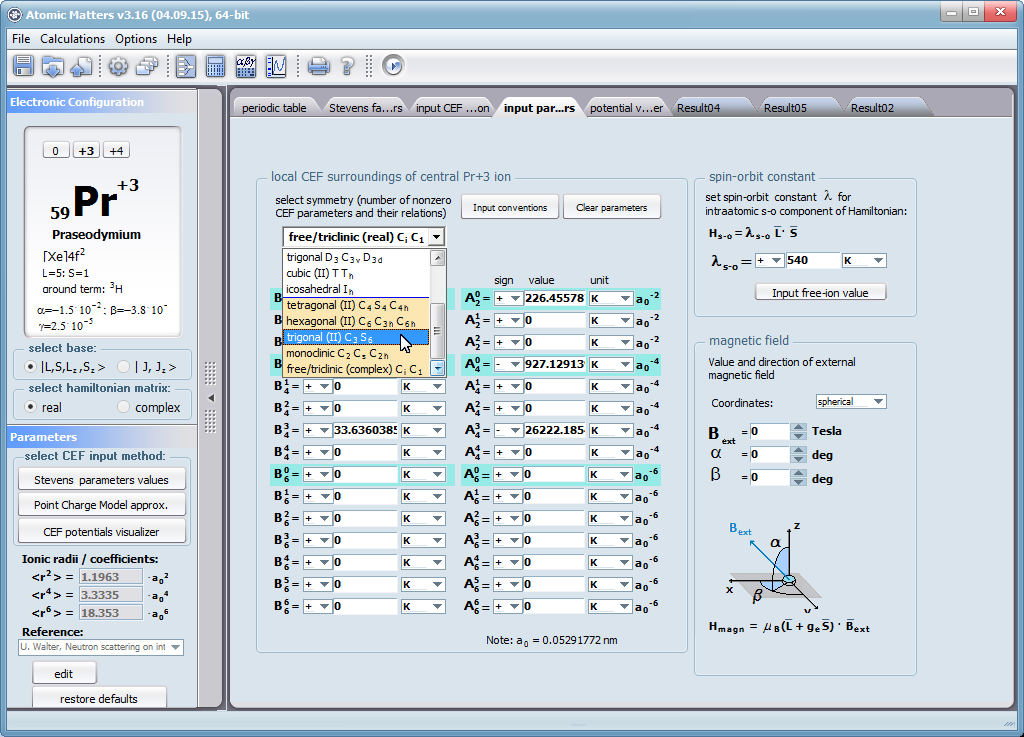
Przedstawiamy zatem aplikację pozwalającą jasno, ściśle i poprawnie teoretycznie zaaplikować fizykę atomu/jonu poddanemu wpływowi pola elektrostatycznego zgodnie z koncepcją Pola Krystalicznego (CEF-Crystal Electric Field, w formalizmie operatorów równoważnych Stevensa, bezpośrednio definiowanym przez twierdzenie Wignera-Eckarta), o współczynnikach definiowanego na wiele sposobów (w tym poprzez model ładunków punktowych, lub wizualizację interaktywną w trzech wymiarach-3D). W każdym miejscu wyboru metod lub zastosowania ewentualnych przybliżeń, aplikacja ustawi wybór domyślny na standard unawany przez d.f.c.s dla danego jonu/atomu lub klasy zagadnień. Dla wartości stałych atomowych używanych do obliczeń masz możliwość przyjęcia parametrów domyślnych – tablicowych lub urealnionych dla typowych sytuacji rachunkowych (których pochodzenie możesz dokumentować i weryfikować), możesz jednak prowadzić rachunki z pełną dowolnością – dobór metod i wartości parametrów jest całkowicie otwarty. Dbając o pełną kontrolowalność obliczeń dajemy pełną władzę nad wprowadzanymi stałymi, których wartość można edytować w zewnętrznych plikach programu wraz z notatką o pochodzeniu wartości edytowanej stałej. Możliwość wglądu wartości składowych elementów wygenerowanej macierzy hamiltonianów dla zadanych parametrów wejściowych pozwala porównywać pełne komplety wynikowych danych z wielu (do 99) obliczeń a każda zależność przedstawiana na wykresie może być uzupełniana danymi porównawczymi (np. eksperymentalnymi, literaturowymi) wprowadzanymi wprost z klawiatury lub poprzez schowek do wygodnego, rozwijalnego arkusza ukrytego po prawej stronie, na każdej zakładce zawierającej wizualizacje temperaturową prezentowanej wielkości. Celem uniknięcia jakichkolwiek problemów natury metrologicznej aplikację wyposażono w wygodny kalkulator jednostek i wielkości, wykonany w postaci układu zakładek tematycznych, pozwalający szybko przeliczyć jednostki używane we wszystkich obliczeniach i definicjach pomiędzy większością znanych standardów. Szereg narzędzi zintegrowanych z aplikacją pozwala na symulacje, transformacje, przeliczenia, porównania i wizualizacje wielu właściwości będących kanonem (modelowym punktem odniesienia) klasycznej fizyki ciała stałego takie jak sieciowe ciepło właściwe Debye’a, wielopoziomowe ciepło elektronowe typu Schottky’ego, entropię statystyczną, prawo Curie i wiele innych.
JAKIE WŁAŚCIWOŚCI MATERII MOŻNA PRZEWIDYWAĆ DZIĘKI ATOMIC MATTERS?
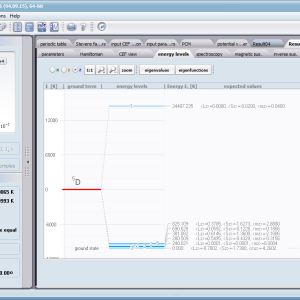
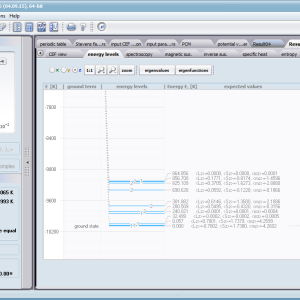
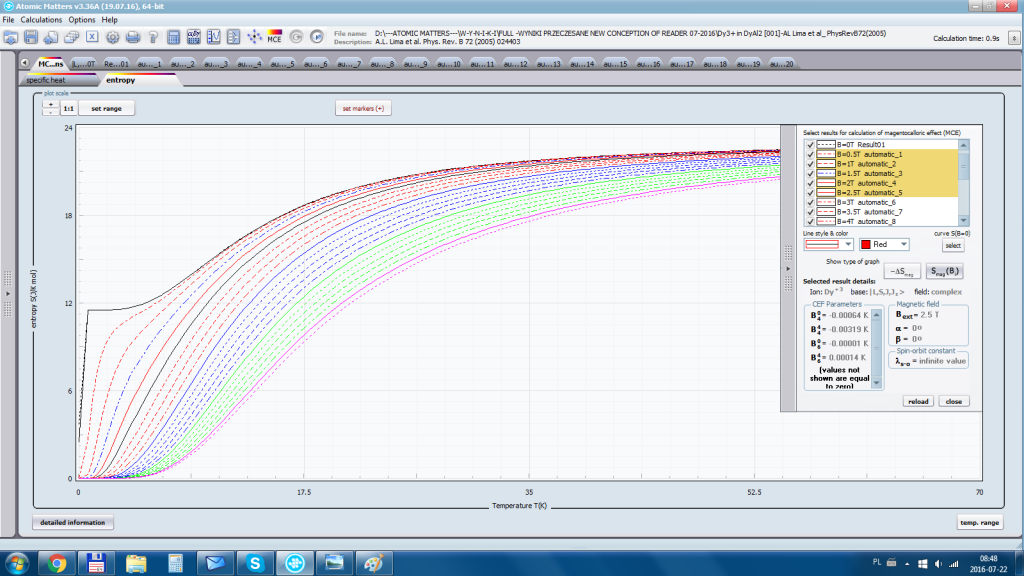
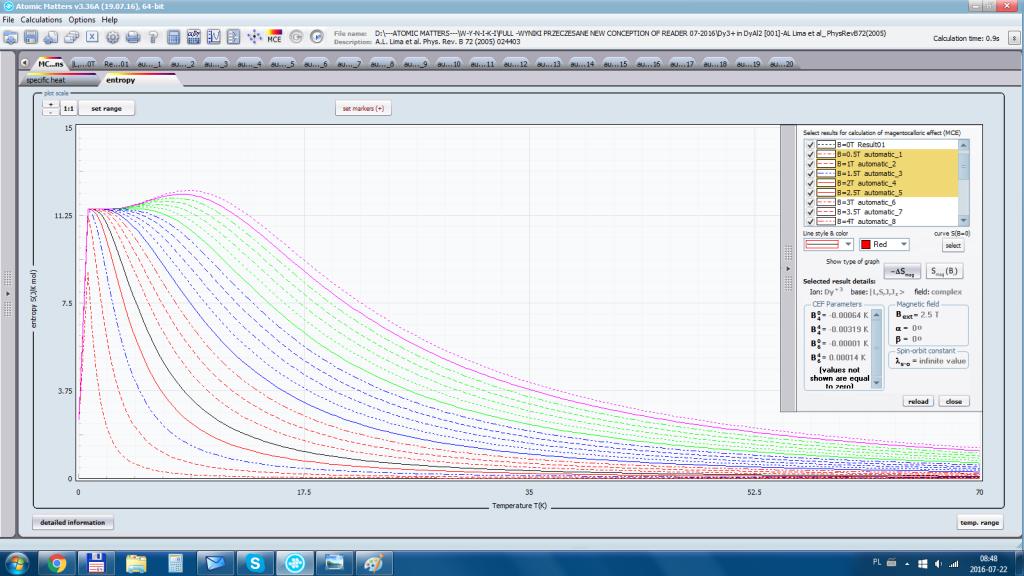
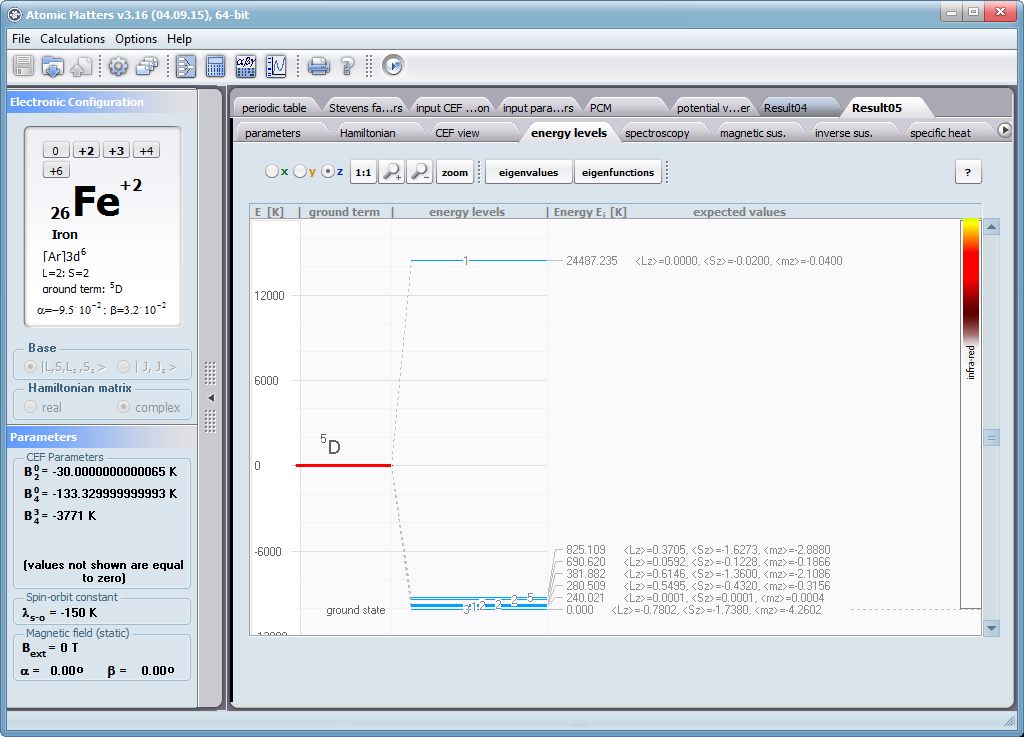
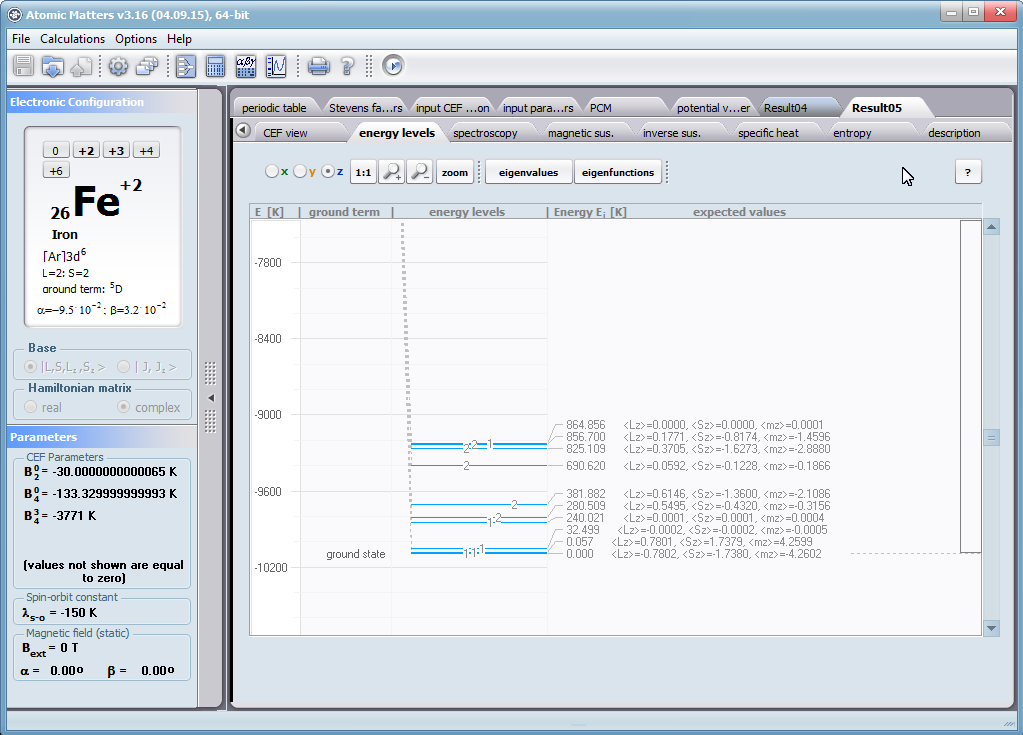
Aplikacja ATOMIC MATTERS umożliwia symulowanie jednojonowych właściwości kryształu, obliczanych ściśle, w oparciu o właściwości elektronowych układów zlokalizowanych, z pełną kontrolą parametrów i metod obliczeniowych. W konsekwencji ATOMIC MATTERS pozwola określić ilościowo właściwości makroskopowe realnych, fizycznie wytwarzalnych lub istniejących materiałów krystalicznych lub prognozować właściwości kryształów obecnie niesyntetyzowanych. Przyjęte metody oraz techniki obliczeń i wizualizacji umożliwią określenie pełnego trójwymiarowego (3D) rozkładu właściwości magnetycznych (podatności magnetycznej, momentu efektywnego, składowych spinowych i orbitalnych, wartości oczekiwanych momentów magnetycznych w stanach i ich komponentów L,S,J), właściwości spektralnych (układ stanów, funkcje własne, macierz transferu prawdopodobieństwa przejść itp.), właściwości kalorymetrycznych (ciepło właściwe typu Shottky’ego i entropia magnetyczna). Wszystkie powyższe właściwości symulujemy w postaci zależności temperaturowych w przedziale zdefiniowanym przez użytkownika. Oczywiście, informacja o właściwościach w temperaturze T = 0[K], gdzie tylko najniższy poziom struktury stanów jest obsadzony również jest dostępna. Całkowity moment magnetyczny układu w T=0[K] jest równy momentowi dla stanu podstawowego. Obsadzenie stanów w niezerowych temperaturach determinuje atomowe właściwości magnetyczne jonów, wpływając bezpośrednio na makroskopowe, magnetyczne właściwości związku. Na podstawie uzyskiwanej, w konsekwencji powyższych obliczeń, struktury stanów wieloelektronowych w przyjętej bazie (|L,S,J,Jz> lub |L,Lz, S,Sz> z wyborem sugerowanym dla typu jonu) obliczalne są następujące właściwości - jako zależności temperaturowe uzyskiwane w przedziale temperatury zdefiniowanym przez użytkownika, takie jak :
- entropia elektronowa –związana z temperaturowym obsadzaniem stanów wieloelektronowych obliczonej struktury elektronowej jonu lub atomu Se (T)
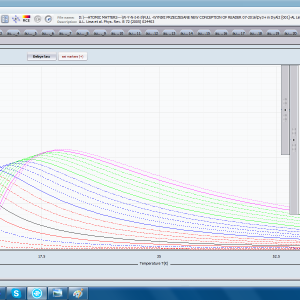
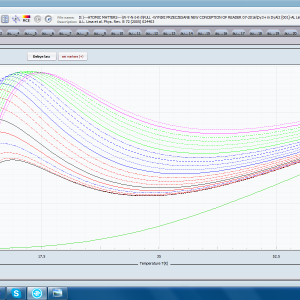
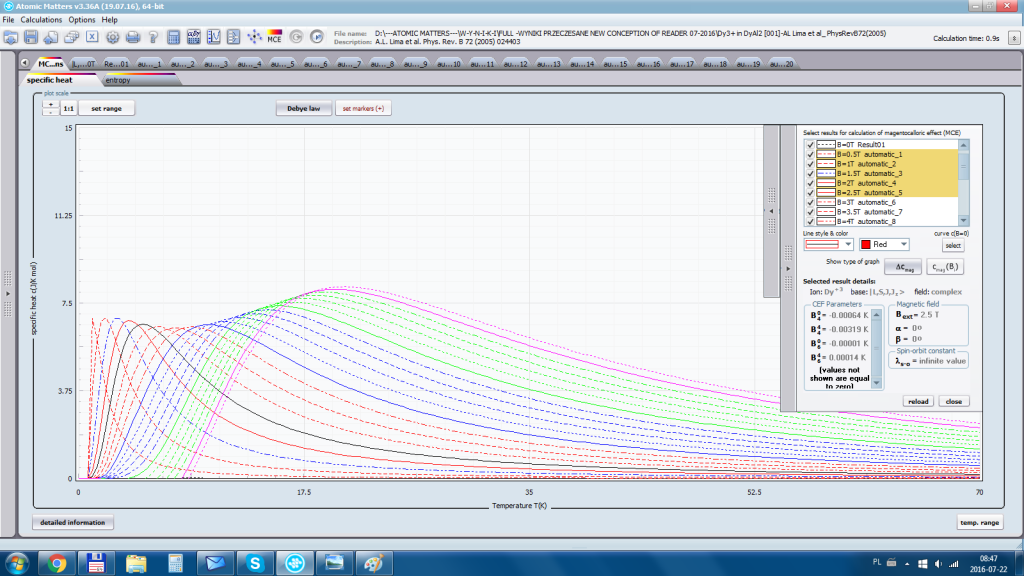
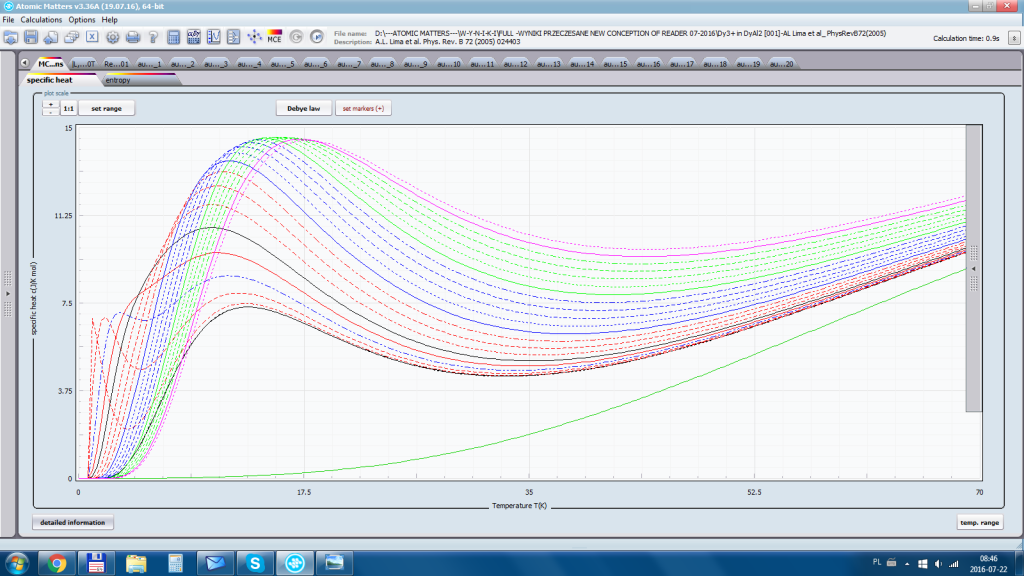
- składowa elektronowa ciepła właściwego – związana z temperaturowym obsadzaniem stanów wieloelektronowych obliczonej struktury elektronowej jonu lub atomu . cSch(T)
- moment magnetyczny i namagnesowanie, oraz jego składowe kierunkowe w zdefiniowanym układzie współrzędnych; mi(B,T)
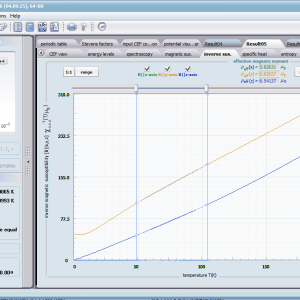
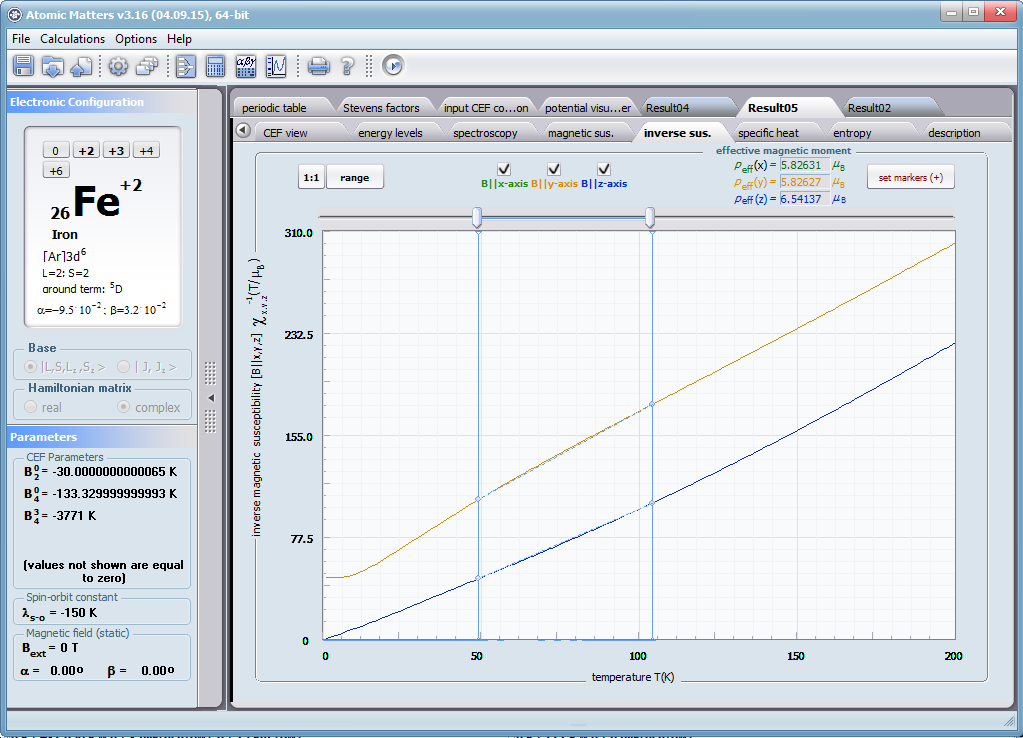
- podatność magnetyczna, jej odwrotność, składowe kierunkowe oraz moment efektywny; χi(T)
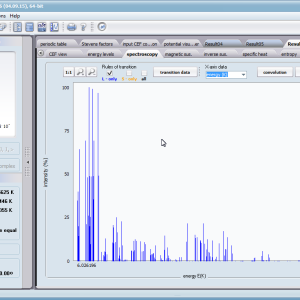
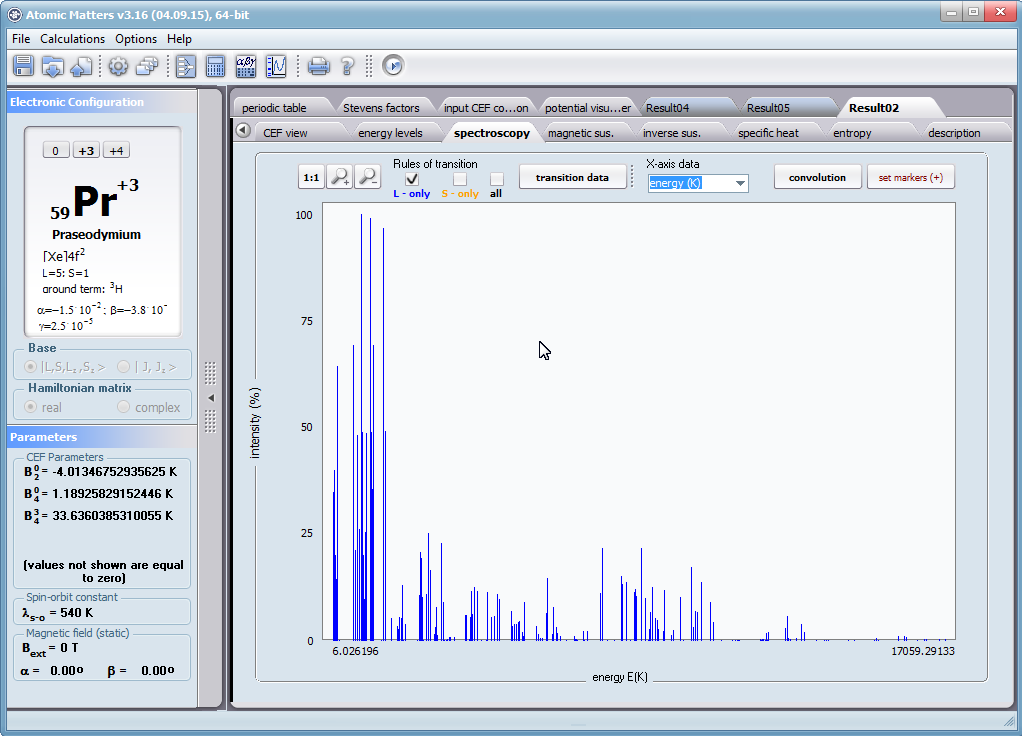
- spektroskopowa obserwowalność przejść międzystanowych i symulowane krzywe absorbcji/emisji dla modelowanych metod spektroskopowych, konwolucje; <Γi|J_|Γj>, <Γi|J+|Γj> , <Γi|JZ|Γj>
- spinowa i orbitalna składowa całkowitego momentu magnetycznego; <Γi|L|Γi>, <Γi|S|Γi>


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}